
Advanced System Building in HTMD#
PDB format considerations#
The PDB format is very old. In an effort to handle its legacy shortcomings, several versions have been made over the years, they are not all readily interchangeable, and not all software can handle each version perfectly. The most important things to watch out for are:
Columns: the PDB format has very rigid rules about what values can go in each space. Keep in mind that it is not a space/tab/comma delimited format, but rather has rigid definitions of what should be in each space/column.
The PDB format as originally designed cannot handle more than 9,999 resids or 99,999 atoms (due to the column format issue). Several workarounds have been devised, such as using hexadecimal numbers or other compact number formats. VMD has no trouble saving more atoms/residues.
Physical and chemical considerations#
One needs to know well the working system, thus:
Always review your PDB file: inspect the REMARK sections of the PDB file. You can often find key specific information regarding the structure (e.g. disulphide bonds, missing atoms, etc.).
Disulphide bonds present in the system must be identified. This is automatically done by HTMD in both CHARMM and Amber.
Metalloproteins: if the metal ion is not an active part of an interaction it may be acceptable to just allow it to act as a cation perhaps restraining it with some harmonic constraints if necessary.
Duplicate atoms in the PDB file: typically simply delete one of the duplicated groups. However, if both conformations are potentially important (e.g. such loops involved in molecular recognition) it might be necessary to simulate both conformations separately.
Protonation/pH#
The protonation state of the system is critical. Since MD simulations typically don’t allow for bond breaking, the initial protonation of the system must be accurate. Knowing what pH you are trying to reproduce is therefore important to obtain the correct results. If you suspect changing protonation is important to your system and you still want to use classical mechanics, consider simulating both states (protonated and not protonated).
Histidine residues can have three different protonations states even at pH 7, therefore, a correct protonation of this residue is particularly critical. This residue can be protonated at either delta (most common; HSD/HID), epsilon (very common also; HSE/HIE) or at both nitrogens (special situations and low pH; HSP/HIP).
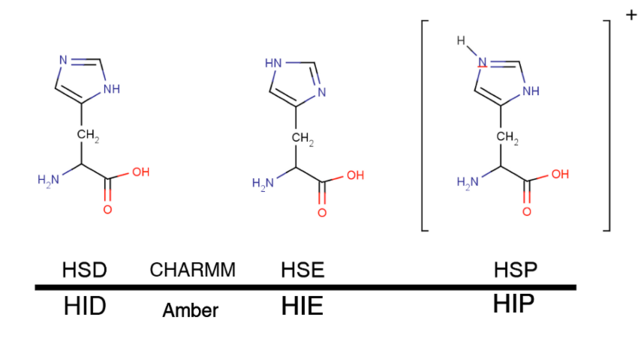
The best way to determine how histidine should be protonated is to look at the the structure. Typically, a histidine residue is protonated if it is close enough to an electron donor (e.g. a glutamic acid), thus creating a hydrogen bond. Since histidines are frequently present at protein active sites, a correct protonation state is particularly important in ligand binding simulations.
In HTMD, one can use systemPrepare to help with protonation.
List of useful tools#
|
Building functions |
|---|---|
— |
List of common CHARMM patches#
C-terminal patches:
Name |
Charge |
Description |
|---|---|---|
CTER |
-1 |
standard C-terminus |
CT1 |
0 |
methylated C-terminus from methyl acetate |
CT2 |
0 |
amidated C-terminus |
CT3 |
0 |
N-Methylamide C-terminus |
N-terminal patches:
Name |
Charge |
Description |
|---|---|---|
NTER |
+1 |
standard N-terminus |
ACE |
0 |
acetylated N-terminus (to create dipeptide) |
ACP |
0 |
acetylated N-terminus (for proline dipeptide) |
PROP |
+1 |
Proline N-Terminal |
GLYP |
+1 |
Glycine N-terminus |
Side-chain patches
Name |
Charge |
Description |
|---|---|---|
ASPP |
0 |
patch for protonated aspartic acid, proton on OD2 |
GLUP |
0 |
patch for protonated glutamic acid, proton on OE2 |
CYSD |
-1 |
patch for deprotonated CYS |
DISU |
+1 |
patch for disulfides. Patch must be 1-CYS and 2-CYS |
HS2 |
+1 |
patch for neutral His, move proton from ND1 to NE2 |
TP1 |
-1 |
patch to convert tyrosine to monoanionic phosphotyrosine |
TP1A |
-1 |
patch to convert tyrosine to monoanionic phenol-phosphate model compound when generating tyr, use first none last none for terminal patches |
TP2 |
-2 |
patch to convert tyrosine to dianionic phosphotyrosine |
TP2A |
-2 |
patch to convert tyrosine to dianionic phosphotyrosine when generating tyr, use first none last none for terminal patches this converts a single tyrosine to a phenol phosphate |
TMP1 |
-1 |
patch to convert tyrosine to monoanionic phosphonate ester O -> methylene (see RESI BMPH) |
TMP2 |
-2 |
patch to convert tyrosine to dianionic phosphonate ester O -> methylene (see RESI BMPD) |
TDF1 |
-1 |
patch to convert tyrosine to monoanionic difluoro phosphonate ester O -> methylene (see RESI BDFH) |
Circular protein chain patches:
Name |
Charge |
Description |
|---|---|---|
LIG1 |
0 |
linkage for cyclic peptide, 1 refers to the C terminus which is a glycine , 2 refers to the N terminus |
LIG2 |
0 |
linkage for cyclic peptide, 1 refers to the C terminus, 2 refers to the N terminus which is a glycine |
LIG3 |
0 |
linkage for cyclic peptide, 1 refers to the C terminus which is a glycine, 2 refers to the N terminus which is a glycine |