
System building: Protein with Ligand#
In this tutorial, we will showcase how to build a protein-ligand system for simulating ligand binding. The sample system is Trypsin (the protein) and benzamidine (the ligand).
Let’s start with some helpful imports and definitions:
from htmd.ui import *
from htmd.home import home
from os.path import join
from moleculekit.config import config
config(viewer='webgl')
datadir = home(dataDir='building-protein-ligand')
2024-06-11 14:58:30,793 - numexpr.utils - INFO - Note: NumExpr detected 20 cores but "NUMEXPR_MAX_THREADS" not set, so enforcing safe limit of 8.
2024-06-11 14:58:30,794 - numexpr.utils - INFO - NumExpr defaulting to 8 threads.
2024-06-11 14:58:30,900 - rdkit - INFO - Enabling RDKit 2022.09.1 jupyter extensions
Please cite HTMD: Doerr et al.(2016)JCTC,12,1845. https://dx.doi.org/10.1021/acs.jctc.6b00049
HTMD Documentation at: https://software.acellera.com/htmd/
You are on the latest HTMD version (2.3.28+0.g1f64e666d.dirty).
Load the protein-ligand complex#
One can obtain the protein-ligand complex from the PDB database (ID:3PTB).
# One can download it directly from the RCSB servers simply by providing the PDB code
prot = Molecule('3PTB')
prot.view()
NGLWidget()
Clean the structures#
The PDB crystal structure contains the protein as well as water molecules, a calcium ion and a ligand. Here we will start by removing the ligand from the protein Molecule as we will add it later to manipulate it separately.
prot.remove('resname BEN')
2024-06-11 14:58:34,227 - moleculekit.molecule - INFO - Removed 9 atoms. 1692 atoms remaining in the molecule.
array([1630, 1631, 1632, 1633, 1634, 1635, 1636, 1637, 1638], dtype=int32)
Preparing the protein#
In this step, we prepare the protein for simulation by adding hydrogens, setting the protonation states, and optimizing the protein (more details on the protein preparation tutorial):
prot = systemPrepare(prot, pH=7.4)
2024-06-11 14:58:34,332 - moleculekit.tools.preparation - WARNING - Both chains and segments are defined in Molecule.chain / Molecule.segid, however they are inconsistent. Protein preparation will use the chain information.
---- Molecule chain report ----
Chain A:
First residue: ILE 16
Final residue: HOH 809
---- End of chain report ----
2024-06-11 14:58:35,888 - moleculekit.tools.preparation - WARNING - The following residues have not been optimized: CA
2024-06-11 14:58:35,968 - moleculekit.tools.preparation - INFO - Modified residue CYS 22 A to CYX
2024-06-11 14:58:35,969 - moleculekit.tools.preparation - INFO - Modified residue HIS 40 A to HIE
2024-06-11 14:58:35,969 - moleculekit.tools.preparation - INFO - Modified residue CYS 42 A to CYX
2024-06-11 14:58:35,970 - moleculekit.tools.preparation - INFO - Modified residue HIS 57 A to HIP
2024-06-11 14:58:35,970 - moleculekit.tools.preparation - INFO - Modified residue CYS 58 A to CYX
2024-06-11 14:58:35,971 - moleculekit.tools.preparation - INFO - Modified residue HIS 91 A to HID
2024-06-11 14:58:35,971 - moleculekit.tools.preparation - INFO - Modified residue CYS 128 A to CYX
2024-06-11 14:58:35,971 - moleculekit.tools.preparation - INFO - Modified residue CYS 136 A to CYX
2024-06-11 14:58:35,972 - moleculekit.tools.preparation - INFO - Modified residue CYS 157 A to CYX
2024-06-11 14:58:35,972 - moleculekit.tools.preparation - INFO - Modified residue CYS 168 A to CYX
2024-06-11 14:58:35,972 - moleculekit.tools.preparation - INFO - Modified residue CYS 182 A to CYX
2024-06-11 14:58:35,973 - moleculekit.tools.preparation - INFO - Modified residue CYS 191 A to CYX
2024-06-11 14:58:35,973 - moleculekit.tools.preparation - INFO - Modified residue CYS 201 A to CYX
2024-06-11 14:58:35,974 - moleculekit.tools.preparation - INFO - Modified residue CYS 220 A to CYX
2024-06-11 14:58:35,974 - moleculekit.tools.preparation - INFO - Modified residue CYS 232 A to CYX
2024-06-11 14:58:35,976 - moleculekit.tools.preparation - WARNING - Dubious protonation state: the pKa of 3 residues is within 1.0 units of pH 7.4.
2024-06-11 14:58:35,977 - moleculekit.tools.preparation - WARNING - Dubious protonation state: TYR 39 A (pKa= 8.24)
2024-06-11 14:58:35,977 - moleculekit.tools.preparation - WARNING - Dubious protonation state: HIS 57 A (pKa= 7.46)
2024-06-11 14:58:35,978 - moleculekit.tools.preparation - WARNING - Dubious protonation state: ASP 189 A (pKa= 6.49)
Define segments#
To build a system in HTMD, we need to separate the chemical molecules into separate segments. This prevents the builder from accidentally bonding different chemical molecules and allows us to add caps to them.
prot = autoSegment(prot, sel='protein')
prot.set('segid', 'W', sel='water')
prot.set('segid', 'CA', sel='resname CA')
2024-06-11 14:58:36,020 - moleculekit.tools.autosegment - INFO - Created segment P0 between resid 16 and 245.
Center the protein to the origin
prot.center()
Let’s work on the ligand!#
Load the ligand from the HTMD data directory. Normally to perform this step you will first need to parameterize your ligand for the AMBER forcefield with a tool like Parameterize (available on PlayMolecule) which produces a topology (cif/mol2) and a parameter (frcmod) file. Here for the sake of the tutorial we provide both files in your HTMD installation.
ligand = Molecule(join(datadir, 'BEN.cif'))
Let’s center the ligand and visualize it:
ligand.center()
ligand.view()
NGLWidget()
# We can give a convenient segid and resname to the ligand
# The resname should be BEN to match the parameters in the
# rtf and prm files.
ligand.set('segid','L')
ligand.set('resname','BEN')
But the ligand is now located inside the protein… We would like the ligand to be:
At a certain distance from the protein
Rotated randomly, to provide different starting conditions
Let’s randomize the ligand position#
ligand.rotateBy(uniformRandomRotation())
This took care of the ligand rotation around its own center. We still need to position it far from the protein. First, find out the radius of the protein:
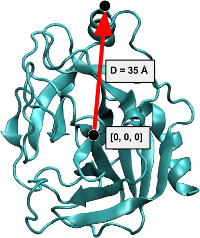
maxdist#
from moleculekit.util import maxDistance
D = maxDistance(prot, 'all')
print(D)
28.830551
D += 10
# Move the ligand 10 Angstrom away from the furthest protein atom in X dimension
ligand.moveBy([D, 0, 0])
# rotateBy rotates by default around [0, 0, 0]. Since the ligand has been moved
# away from the center it will be rotated in a sphere of radius D+10 around [0, 0, 0]
ligand.rotateBy(uniformRandomRotation())
Mix it all together#
mol = Molecule(name='combo')
mol.append(prot)
mol.append(ligand)
mol.reps.add(sel='protein', style='NewCartoon', color='Secondary Structure')
mol.reps.add(sel='resname BEN', style='Licorice')
mol.view()
NGLWidget()
Solvate#
Water is the driving force of all nature. –Leonardo da Vinci
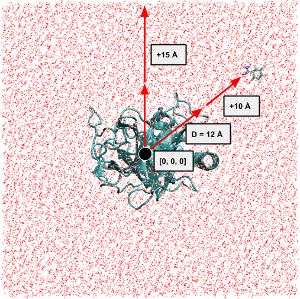
waterbox#
# We solvate with a larger cubic box to fully solvate the ligand
DW = D + 5
smol = solvate(mol, minmax=[[-DW, -DW, -DW], [DW, DW, DW]])
smol.reps.add(sel='water', style='Lines')
smol.view()
2024-06-11 14:58:36,251 - htmd.builder.solvate - INFO - Using water pdb file at: /home/sdoerr/Work/htmd/htmd/share/solvate/wat.pdb
2024-06-11 14:58:36,587 - htmd.builder.solvate - INFO - Replicating 8 water segments, 2 by 2 by 2
Solvating: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 8/8 [00:02<00:00, 3.05it/s]
2024-06-11 14:58:39,744 - htmd.builder.solvate - INFO - 20145 water molecules were added to the system.
NGLWidget()
Build the System with a specific forcefield#
HTMD aims to be force-field agnostic. After you have a built system, you can either build it in Amber or CHARMM. The following sections work on the same previously solvated system and can be interconverted. However due to licensing issues we are not able to provide CHARMM support and recommend using the AMBER forcefield.
Special care must be taken care in this case due to the use of benzamidine, which is not present by default on the respective forcefields.
CHARMM forcefield#
charmm.listFiles()
---- Topologies files list: /home/sdoerr/Work/htmd/htmd/share/builder/charmmfiles/top/ ----
/top/top_all22_prot.rtf
/top/top_all22star_prot.rtf
/top/top_all35_ethers.rtf
/top/top_all36_carb.rtf
/top/top_all36_cgenff.rtf
/top/top_all36_lipid.rtf
/top/top_all36_lipid_ljpme.rtf
/top/top_all36_na.rtf
/top/top_all36_prot.rtf
/top/top_water_ions.rtf
---- Parameters files list: /home/sdoerr/Work/htmd/htmd/share/builder/charmmfiles/par/ ----
/par/par_all22_prot.prm
/par/par_all22star_prot.prm
/par/par_all35_ethers.prm
/par/par_all36_carb.prm
/par/par_all36_cgenff.prm
/par/par_all36_lipid.prm
/par/par_all36_lipid_ljpme.prm
/par/par_all36_na.prm
/par/par_all36m_prot.prm
/par/par_water_ions.prm
---- Stream files list: /home/sdoerr/Work/htmd/htmd/share/builder/charmmfiles/str/ ----
/str/carb/toppar_all36_carb_glycolipid.str
/str/carb/toppar_all36_carb_glycopeptide.str
/str/carb/toppar_all36_carb_imlab.str
/str/carb/toppar_all36_carb_lignin.str
/str/carb/toppar_all36_carb_model.str
/str/cphmd/protpatch_protein_toppar36.str
/str/lipid/toppar_all36_lipid_bacterial.str
/str/lipid/toppar_all36_lipid_bacterial_ljpme.str
/str/lipid/toppar_all36_lipid_cardiolipin.str
/str/lipid/toppar_all36_lipid_cationpi_wyf.str
/str/lipid/toppar_all36_lipid_cholesterol.str
/str/lipid/toppar_all36_lipid_cholesterol_model_1.str
/str/lipid/toppar_all36_lipid_detergent.str
/str/lipid/toppar_all36_lipid_ether.str
/str/lipid/toppar_all36_lipid_ether_ljpme.str
/str/lipid/toppar_all36_lipid_inositol.str
/str/lipid/toppar_all36_lipid_list.str
/str/lipid/toppar_all36_lipid_lps.str
/str/lipid/toppar_all36_lipid_miscellaneous.str
/str/lipid/toppar_all36_lipid_miscellaneous_ljpme.str
/str/lipid/toppar_all36_lipid_model.str
/str/lipid/toppar_all36_lipid_prot.str
/str/lipid/toppar_all36_lipid_sphingo.str
/str/lipid/toppar_all36_lipid_yeast.str
/str/lipid/toppar_all36_lipid_yeast_ljpme.str
/str/misc/toppar_amines.str
/str/misc/toppar_cgenff_misc.str
/str/misc/toppar_dum_noble_gases.str
/str/misc/toppar_hbond.str
/str/misc/toppar_ions_won.str
/str/na/toppar_all36_na_model.str
/str/na/toppar_all36_na_modifications.str
/str/na/toppar_all36_na_nad_ppi.str
/str/na/toppar_all36_na_reactive_rna.str
/str/na/toppar_all36_na_rna_modified.str
/str/na/toppar_all36_na_rna_modified_for_new_psf_gen_code_2022.str
/str/prot/toppar_all36_prot_aldehydes.str
/str/prot/toppar_all36_prot_arg0.str
/str/prot/toppar_all36_prot_c36m_d_aminoacids.str
/str/prot/toppar_all36_prot_fluoro_alkanes.str
/str/prot/toppar_all36_prot_heme.str
/str/prot/toppar_all36_prot_heme_for_new_psf_gen_code_2022.str
/str/prot/toppar_all36_prot_model.str
/str/prot/toppar_all36_prot_modify_res.str
/str/prot/toppar_all36_prot_na_combined.str
/str/prot/toppar_all36_prot_pyridines.str
/str/prot/toppar_all36_prot_retinol.str
Build and ionize using CHARMM#
topos_charmm = charmm.defaultTopo() + [join(datadir, 'BEN.rtf')]
params_charmm = charmm.defaultParam() + [join(datadir, 'BEN.prm')]
# bmol_charmm = charmm.build(smol, topo=topos_charmm, param=params_charmm, outdir='./build_charmm')
AMBER forcefield#
amber.listFiles()
---- Forcefield files list: /home/sdoerr/miniforge3/envs/htmd/dat/leap/cmd/ ----
leaprc.amberdyes
leaprc.conste
leaprc.constph
leaprc.DNA.bsc1
leaprc.DNA.OL15
leaprc.ffAM1
leaprc.ffPM3
leaprc.fluorine.ff15ipq
leaprc.gaff
leaprc.gaff2
leaprc.GLYCAM_06EPb
leaprc.GLYCAM_06j-1
leaprc.lipid21
leaprc.mimetic.ff15ipq
leaprc.modrna08
leaprc.music
leaprc.phosaa10
leaprc.phosaa14SB
leaprc.phosaa19SB
leaprc.protein.fb15
leaprc.protein.ff03.r1
leaprc.protein.ff03ua
leaprc.protein.ff14SB
leaprc.protein.ff14SB_modAA
leaprc.protein.ff14SBonlysc
leaprc.protein.ff15ipq
leaprc.protein.ff15ipq-vac
leaprc.protein.ff19ipq
leaprc.protein.ff19SB
leaprc.protein.ff19SB_modAA
leaprc.RNA.LJbb
leaprc.RNA.OL3
leaprc.RNA.ROC
leaprc.RNA.Shaw
leaprc.RNA.YIL
leaprc.water.fb3
leaprc.water.fb4
leaprc.water.opc
leaprc.water.opc3
leaprc.water.opc3pol
leaprc.water.spce
leaprc.water.spceb
leaprc.water.tip3p
leaprc.water.tip4pd
leaprc.water.tip4pd-a99SBdisp
leaprc.water.tip4pew
leaprc.xFPchromophores
---- OLD Forcefield files list: /home/sdoerr/miniforge3/envs/htmd/dat/leap/cmd/ ----
oldff/leaprc.DNA.bsc0
oldff/leaprc.ff02
oldff/leaprc.ff02pol.r0
oldff/leaprc.ff02pol.r1
oldff/leaprc.ff02polEP.r0
oldff/leaprc.ff02polEP.r1
oldff/leaprc.ff03
oldff/leaprc.ff10
oldff/leaprc.ff14ipq
oldff/leaprc.ff14SB
oldff/leaprc.ff14SB.redq
oldff/leaprc.ff84
oldff/leaprc.ff86
oldff/leaprc.ff94
oldff/leaprc.ff94.nmr
oldff/leaprc.ff96
oldff/leaprc.ff98
oldff/leaprc.ff99
oldff/leaprc.ff99bsc0
oldff/leaprc.ff99SB
oldff/leaprc.ff99SBildn
oldff/leaprc.ff99SBnmr
oldff/leaprc.GLYCAM_04
oldff/leaprc.GLYCAM_06
oldff/leaprc.GLYCAM_06EP
oldff/leaprc.GLYCAM_06h
oldff/leaprc.GLYCAM_06h-1
oldff/leaprc.GLYCAM_06h-12SB
oldff/leaprc.GLYCAM_06j_10
oldff/leaprc.lipid11
oldff/leaprc.lipid14
oldff/leaprc.lipid17
oldff/leaprc.parmbsc0_chiOL4_ezOL1
oldff/leaprc.rna.ff02
oldff/leaprc.rna.ff02EP
oldff/leaprc.rna.ff84
oldff/leaprc.rna.ff94
oldff/leaprc.rna.ff98
oldff/leaprc.rna.ff99
oldff/leaprc.toyrna
---- Topology files list: /home/sdoerr/miniforge3/envs/htmd/dat/leap/prep/ ----
all_amino03.in
all_aminoct03.in
all_aminont03.in
amino10.in
amino12.in
aminoct10.in
aminoct12.in
aminont10.in
aminont12.in
chcl3.in
dna_nuc94-bsc0_chiOl4-ezOL1.in
epACE.prepin
epALA.prepin
epARG.prepin
epASN.prepin
epASP.prepin
epCYS.prepin
epGLN.prepin
epGLU.prepin
epGLY.prepin
epHID.prepin
epHIE.prepin
epILE.prepin
epLEU.prepin
epLYS.prepin
epMET.prepin
epNME.prepin
epPHE.prepin
epSER.prepin
epTHR.prepin
epTRP.prepin
epTYR.prepin
epVAL.prepin
GLYCAM_06EPb.prep
GLYCAM_06j-1.prep
GLYCAM_lipids_06h.prep
meoh.in
nma.in
nucleic10.in
toyrna.in
uni_amino03.in
uni_aminoct03.in
uni_aminont03.in
---- Parameter files list: /home/sdoerr/miniforge3/envs/htmd/dat/leap/parm/ ----
frcmod.chcl3
frcmod.chiOL4
frcmod.conste
frcmod.constph
frcmod.dc4
frcmod.DNA.OL15
frcmod.fb15
frcmod.ff02pol.r1
frcmod.ff03
frcmod.ff03ua
frcmod.ff12SB
frcmod.ff14SB
frcmod.ff14SBmodAA
frcmod.ff15ipq-19F
frcmod.ff15ipq-m
frcmod.ff19SB
frcmod.ff19SB_XXX
frcmod.ff19SBmodAA
frcmod.ff99bsc0CG
frcmod.ff99SB
frcmod.ff99SB14
frcmod.ff99SBildn
frcmod.ff99SBnmr
frcmod.ff99SP
frcmod.ions1lm_1264_spce
frcmod.ions1lm_1264_tip3p
frcmod.ions1lm_1264_tip4pew
frcmod.ions1lm_126_spce
frcmod.ions1lm_126_tip3p
frcmod.ions1lm_126_tip4pew
frcmod.ions1lm_iod
frcmod.ions234lm_1264_spce
frcmod.ions234lm_1264_tip3p
frcmod.ions234lm_1264_tip4pew
frcmod.ions234lm_126_spce
frcmod.ions234lm_126_tip3p
frcmod.ions234lm_126_tip4pew
frcmod.ions234lm_hfe_spce
frcmod.ions234lm_hfe_tip3p
frcmod.ions234lm_hfe_tip4pew
frcmod.ions234lm_iod_spce
frcmod.ions234lm_iod_tip3p
frcmod.ions234lm_iod_tip4pew
frcmod.ions_charmm22
frcmod.ionsff99_tip3p
frcmod.ionsjc_spce
frcmod.ionsjc_tip3p
frcmod.ionsjc_tip4pew
frcmod.ionslm_1264_fb3
frcmod.ionslm_1264_fb4
frcmod.ionslm_1264_opc
frcmod.ionslm_1264_opc3
frcmod.ionslm_126_fb3
frcmod.ionslm_126_fb4
frcmod.ionslm_126_opc
frcmod.ionslm_126_opc3
frcmod.ionslm_hfe_fb3
frcmod.ionslm_hfe_fb4
frcmod.ionslm_hfe_opc
frcmod.ionslm_hfe_opc3
frcmod.ionslm_iod_fb3
frcmod.ionslm_iod_fb4
frcmod.ionslm_iod_opc
frcmod.ionslm_iod_opc3
frcmod.meoh
frcmod.nma
frcmod.opc
frcmod.opc3
frcmod.opc3pol
frcmod.opc3pol_HMR4fs
frcmod.parmbsc0
frcmod.parmbsc0_ezOL1
frcmod.parmbsc1
frcmod.parmCHI_YIL
frcmod.phmd
frcmod.phosaa10
frcmod.phosaa14SB
frcmod.phosaa19SB
frcmod.pol3
frcmod.protonated_nucleic
frcmod.qspcfw
frcmod.RNA.LJbb
frcmod.ROC-RNA
frcmod.ROC-RNA_const
frcmod.shaw
frcmod.spce
frcmod.spceb
frcmod.spcfw
frcmod.tip3p
frcmod.tip3pf
frcmod.tip3pfb
frcmod.tip4p
frcmod.tip4pd
frcmod.tip4pd-a99SB-disp
frcmod.tip4pew
frcmod.tip4pfb
frcmod.tip5p
frcmod.urea
frcmod.vdWall
frcmod.xFPchromophores
frcmod.xFPchromophores.2016
frcmod.xFPchromophores.2022
---- Extra forcefield files list: /home/sdoerr/Work/htmd/htmd/share/builder/amberfiles/ ----
ff-nucleic-OL15/leaprc.ff-nucleic-OL15
---- Extra topology files list: /home/sdoerr/Work/htmd/htmd/share/builder/amberfiles/ ----
cofactors/cofactors.in
ff-ncaa/ffncaa.in
ff-nucleic-OL15/leap-ff-nucleic-OL15.in
ff-ptm/ffptm.in
---- Extra parameter files list: /home/sdoerr/Work/htmd/htmd/share/builder/amberfiles/ ----
cofactors/ADP.frcmod
cofactors/AMP.frcmod
cofactors/ATP.frcmod
cofactors/FMN.frcmod
cofactors/GDP.frcmod
cofactors/GTP.frcmod
cofactors/HEM.frcmod
cofactors/NAD.frcmod
cofactors/NAI.frcmod
cofactors/NAP.frcmod
ff-ncaa/004.frcmod
ff-ncaa/03Y.frcmod
ff-ncaa/0A1.frcmod
ff-ncaa/0BN.frcmod
ff-ncaa/1MH.frcmod
ff-ncaa/2AS.frcmod
ff-ncaa/2GX.frcmod
ff-ncaa/2ML.frcmod
ff-ncaa/4IN.frcmod
ff-ncaa/4PH.frcmod
ff-ncaa/5JP.frcmod
ff-ncaa/AA4.frcmod
ff-ncaa/ABA.frcmod
ff-ncaa/AHP.frcmod
ff-ncaa/ALC.frcmod
ff-ncaa/ALN.frcmod
ff-ncaa/APD.frcmod
ff-ncaa/BB8.frcmod
ff-ncaa/BCS.frcmod
ff-ncaa/CCS.frcmod
ff-ncaa/CSA.frcmod
ff-ncaa/D4P.frcmod
ff-ncaa/DAB.frcmod
ff-ncaa/DPP.frcmod
ff-ncaa/ESC.frcmod
ff-ncaa/FGL.frcmod
ff-ncaa/GHG.frcmod
ff-ncaa/GME.frcmod
ff-ncaa/GNC.frcmod
ff-ncaa/HHK.frcmod
ff-ncaa/HLU.frcmod
ff-ncaa/HLX.frcmod
ff-ncaa/HOX.frcmod
ff-ncaa/HPE.frcmod
ff-ncaa/HQA.frcmod
ff-ncaa/HTR.frcmod
ff-ncaa/I2M.frcmod
ff-ncaa/IGL.frcmod
ff-ncaa/IIL.frcmod
ff-ncaa/IML.frcmod
ff-ncaa/KYN.frcmod
ff-ncaa/LME.frcmod
ff-ncaa/LMQ.frcmod
ff-ncaa/ME0.frcmod
ff-ncaa/MEA.frcmod
ff-ncaa/MEN.frcmod
ff-ncaa/MEQ.frcmod
ff-ncaa/MLE.frcmod
ff-ncaa/MLZ.frcmod
ff-ncaa/MME.frcmod
ff-ncaa/MMO.frcmod
ff-ncaa/MVA.frcmod
ff-ncaa/NAL.frcmod
ff-ncaa/NCY.frcmod
ff-ncaa/NLE.frcmod
ff-ncaa/NVA.frcmod
ff-ncaa/NZC.frcmod
ff-ncaa/OCY.frcmod
ff-ncaa/OMX.frcmod
ff-ncaa/ONL.frcmod
ff-ncaa/TRO.frcmod
ff-ncaa/TY2.frcmod
ff-ncaa/TYQ.frcmod
ff-ncaa/YCM.frcmod
ff-ncaa/YNM.frcmod
ff-nucleic-OL15/ff-nucleic-OL15.frcmod
ff-ptm/0AF.frcmod
ff-ptm/2MR.frcmod
ff-ptm/4PQ.frcmod
ff-ptm/ALY.frcmod
ff-ptm/BTK.frcmod
ff-ptm/CGU.frcmod
ff-ptm/CSO.frcmod
ff-ptm/CSP.frcmod
ff-ptm/CSS.frcmod
ff-ptm/DA2.frcmod
ff-ptm/DAH.frcmod
ff-ptm/HY3.frcmod
ff-ptm/HYP.frcmod
ff-ptm/LYZ.frcmod
ff-ptm/M3L.frcmod
ff-ptm/MLY.frcmod
ff-ptm/ORM.frcmod
ff-ptm/P1L.frcmod
ff-ptm/PCA.frcmod
ff-ptm/PRK.frcmod
ff-ptm/PTR.frcmod
ff-ptm/SEP.frcmod
ff-ptm/TPO.frcmod
Build and ionize using Amber#
topos_amber = amber.defaultTopo() + [join(datadir, 'BEN.cif')]
frcmods_amber = amber.defaultParam() + [join(datadir, 'BEN.frcmod')]
bmol_amber = amber.build(smol, topo=topos_amber, param=frcmods_amber, outdir='./build_amber', saltconc=0.15)
2024-06-11 14:58:44,189 - htmd.builder.amber - INFO - Detecting disulfide bonds.
2024-06-11 14:58:44,197 - htmd.builder.builder - INFO - 6 disulfide bonds were added
Disulfide Bond between: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 26, insertion: '', segid: 'P0'>
and: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 42, insertion: '', segid: 'P0'>
Disulfide Bond between: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 117, insertion: '', segid: 'P0'>
and: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 184, insertion: '', segid: 'P0'>
Disulfide Bond between: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 174, insertion: '', segid: 'P0'>
and: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 198, insertion: '', segid: 'P0'>
Disulfide Bond between: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 149, insertion: '', segid: 'P0'>
and: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 163, insertion: '', segid: 'P0'>
Disulfide Bond between: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 8, insertion: '', segid: 'P0'>
and: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 138, insertion: '', segid: 'P0'>
Disulfide Bond between: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 110, insertion: '', segid: 'P0'>
and: UniqueResidueID<resname: 'CYX', chain: 'A', resid: 211, insertion: '', segid: 'P0'>
2024-06-11 14:58:45,604 - htmd.builder.amber - INFO - Starting the build.
2024-06-11 14:58:48,790 - htmd.builder.amber - INFO - Finished building.
2024-06-11 14:58:49,566 - moleculekit.writers - WARNING - Field "resid" of PDB overflows. Your data will be truncated to 4 characters.
2024-06-11 14:58:54,542 - htmd.builder.ionize - INFO - Adding 10 anions + 0 cations for neutralizing and 0 ions for the given salt concentration 0 M.
2024-06-11 14:58:57,279 - htmd.builder.amber - INFO - Starting the build.
2024-06-11 14:59:00,594 - htmd.builder.amber - INFO - Finished building.
2024-06-11 14:59:01,416 - moleculekit.writers - WARNING - Field "resid" of PDB overflows. Your data will be truncated to 4 characters.
2024-06-11 14:59:05,984 - py.warnings - WARNING - /home/sdoerr/miniforge3/envs/htmd/lib/python3.10/site-packages/Bio/pairwise2.py:278: BiopythonDeprecationWarning: Bio.pairwise2 has been deprecated, and we intend to remove it in a future release of Biopython. As an alternative, please consider using Bio.Align.PairwiseAligner as a replacement, and contact the Biopython developers if you still need the Bio.pairwise2 module.
warnings.warn(
2024-06-11 14:59:08,752 - moleculekit.tools.sequencestructuralalignment - INFO - Alignment #0 was done on 223 residues: 2-224
Visualize#
The built system can be visualized (with waters hidden to be able to visualize the inserted ions):
bmol_amber.view(sel='not water') # visualize the charmm built system
# bmol_amber.view(sel='not water') # uncomment to visualize the amber built system
2024-06-11 14:59:09,148 - moleculekit.writers - WARNING - Field "resid" of PDB overflows. Your data will be truncated to 4 characters.
NGLWidget()
The bmol_charmm and bmol_amber are Molecule objects that
contain the built system, but the full contents to run a simulation are
located in the outdir (./build in this case).