
System preparation#
What is system preparation?#
The protonation state of a biological system is critical. Since MD simulations typically don’t allow for bond breaking, the initial protonation of the system must be accurate. Knowing what pH you are trying to reproduce and the protonation state of all molecules is therefore important to obtain the correct results. If you suspect changing protonation is important to your system and you still want to use classical mechanics, consider running multiple simulations with different protonation states.
Histidine residues can have three different protonations states even at pH 7, therefore, a correct protonation of this residue is particularly critical. This residue can be protonated at either delta (most common; HSD/HID), epsilon (very common also; HSE/HIE) or at both nitrogens (special situations and low pH; HSP/HIP).
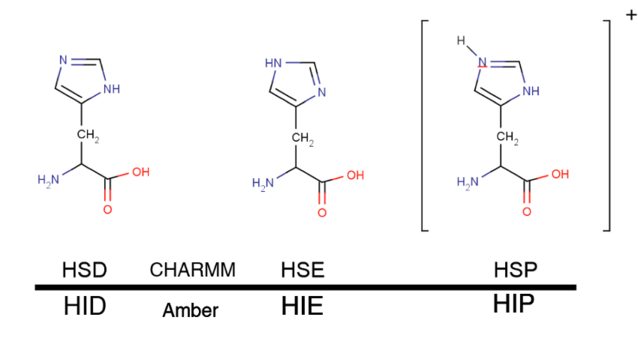
histidines#
The best way to determine how histidine should be protonated is to look at the the structure. Typically, a histidine residue is protonated if it is close enough to an electron donor (e.g. a glutamic acid), thus creating a hydrogen bond. Since histidines are frequently present at protein active sites, a correct protonation state is particularly important in ligand binding simulations.
In HTMD, one can use systemPrepare to help with protonation.
The system preparation phase, based on the PDB2PQR and propKa softwares, addresses e.g. the problems of assigning titration states at the user-chosen pH; flipping the side chains of HIS, ASN, and GLN residues; and optimizing the overall hydrogen bonding network.
After preparing, the build phase takes a prepared system and applies the chosen forcefield in order to obtain simulation-ready input files.
Let’s start#
from htmd.ui import *
config(viewer='ngl')
2021-11-16 09:53:00,399 - numexpr.utils - INFO - NumExpr defaulting to 8 threads.
Please cite HTMD: Doerr et al.(2016)JCTC,12,1845. https://dx.doi.org/10.1021/acs.jctc.6b00049
HTMD Documentation at: https://www.htmd.org/docs/latest/
You are on the latest HTMD version (unpackaged : /home/sdoerr/Work/htmd/htmd).
System Preparation in HTMD#
The system preparation phase is based on the PDB2PQR software. It includes the following steps (from the PDB2PQR algorithm description):
Compute empirical pKa values for the residues’ local environment (propKa)
Assign titration states at the user-chosen pH;
Flipping the side chains of HIS (including user defined HIS states), ASN, and GLN residues;
Rotating the sidechain hydrogen on SER, THR, TYR, and CYS (if available);
Determining the best placement for the sidechain hydrogen on neutral HIS, protonated GLU, and protonated ASP;
Optimizing all water hydrogens.
The hydrogen bonding network calculations are performed by the PDB2PQR software package. The pKa calculations are performed by the PROPKA 3.1 software packages. Please see the copyright, license and citation terms distributed with each.
Note that this version was modified in order to use an externally-supplied propKa 3.1 (installed automatically via dependencies), whereas the original had propKa 3.0 embedded!
The results of the function should be roughly equivalent of the system preparation wizard’s preprocessing and optimization steps of Schrodinger’s Maestro software.
Protein residue pKas in water#

residue_naming#
Modified residue names#
The molecule produced by the preparation modifies residue names according to their protonation. Later system-building functions assume these residue naming conventions.
Charge +1 |
Neutral |
Charge -1 |
|---|---|---|
ASH |
ASP |
|
CYS |
CYM |
|
GLH |
GLU |
|
HIP |
HID/HIE |
|
LYS |
LYN |
|
TYR |
TYM |
|
ARG |
AR0 |
Note: support for alternative charge states varies between the forcefields.
Limitations#
PDB2PQR: returns one solution consistent with its restraints, not the optimal one;
Membrane proteins: propKa ignores lipid exposure (more on this later);
Large conformational changes: local environment changes may be large enough that pKa decisions are not transferable.
systemPrepare function#
The systemPrepare function requires a Molecule object, the
protein/DNA/RNA to be prepared, as an argument, and returns the prepared
system, also as a Molecule. Logging messages will provide
information and warnings about the process.
def systemPrepare(mol_in,
pH=7.0,
verbose=0,
return_details=False,
hydrophobic_thickness=None):
Returns a Molecule object, where residues have been renamed to
follow internal conventions on protonation (below). Coordinates are
changed to optimize the H-bonding network.
Parameters#
help(systemPrepare)
Help on function systemPrepare in module moleculekit.tools.preparation:
systemPrepare(mol_in, titration=True, pH=7.4, force_protonation=None, no_opt=None, no_prot=None, no_titr=None, hold_nonpeptidic_bonds=True, verbose=True, return_details=False, hydrophobic_thickness=None, plot_pka=None, _logger_level='ERROR', _molkit_ff=True)
Prepare molecular systems through protonation and h-bond optimization.
The preparation routine protonates and optimizes protein and nucleic residues.
It will also take into account any non-protein, non-nucleic molecules for the pKa calculation
but will not attempt to protonate or optimize those.
Returns a Molecule object, where residues have been renamed to follow
internal conventions on protonation (below). Coordinates are changed to
optimize the H-bonding network.
The following residue names are used in the returned molecule:
=== ===============================
ASH Neutral ASP
CYX SS-bonded CYS
CYM Negative CYS
GLH Neutral GLU
HIP Positive HIS
HID Neutral HIS, proton HD1 present
HIE Neutral HIS, proton HE2 present
LYN Neutral LYS
TYM Negative TYR
AR0 Neutral ARG
=== ===============================
========= ======= =========
Charge +1 Neutral Charge -1
========= ======= =========
- ASH ASP
- CYS CYM
- GLH GLU
HIP HID/HIE -
LYS LYN -
- TYR TYM
ARG AR0 -
========= ======= =========
A detailed table about the residues modified is returned (as a second return value) when
return_details is True .
If hydrophobic_thickness is set to a positive value 2*h, a warning is produced for titratable residues
having -h<z<h and are buried in the protein by less than 75%. Note that the heuristic for the
detection of membrane-exposed residues is very crude; the "buried fraction" computation
(from propKa) is approximate; also, in the presence of cavities,
residues may be solvent-exposed independently from their z location.
Notes
-----
Features:
- assigns protonation states via propKa
- flips residues to optimize H-bonding network
- debumps collisions
- fills in missing atoms, e.g. hydrogen atoms
Parameters
----------
mol_in : moleculekit.molecule.Molecule
the object to be optimized
titration : bool
If set to True it will use propka to set the titration state of residues. Otherwise it will just add and optimize the hydrogens.
pH : float
pH to decide titration
verbose : bool
verbosity
return_details : bool
whether to return just the prepared Molecule (False, default) or a molecule and a ResidueInfo
object including computed properties
hydrophobic_thickness : float
the thickness of the membrane in which the protein is embedded, or None if globular protein.
Used to provide a warning about membrane-exposed residues.
force_protonation : list of tuples
Allows the user to force specific protonations on residues. This can be done by providing a list of tuples,
one for each residue we want to force. i.e. [("protein and resid 40", "HID")] will force the protonation of
the first atomselection to the second resname. Atomselections should be valid VMD atomselections.
no_opt : list of str
Allows the user to disable optimization for specific residues. For example if the user determines that a
residue flip or change of coordinates performed by this method causes issues in the structure, they can
disable optimization on that residue by passing an atomselection for the residue to hold. i.e. ["protein and resid 23"].
no_prot : list of str
Same as no_opt but disables the addition of hydrogens to specific residues.
no_titr : list of str
Same as no_opt but disables the titration of specific residues.
hold_nonpeptidic_bonds : bool
When set to True, systemPrepare will automatically not optimize, protonate or titrate protein residues
which are covalently bound to non-protein molecules. When set to False, systemPrepare will optimize them
ignoring the covalent bond, meaning it may break the bonds or add hydrogen atoms between the bonds.
plot_pka : str
Provide a file path with .png extension to draw the titration diagram for the system residues.
Returns
-------
mol_out : moleculekit.molecule.Molecule
the molecule titrated and optimized. The molecule object contains an additional attribute,
details : pandas.DataFrame
A table of residues with the corresponding protonation states, pKas, and other information
Examples
--------
>>> tryp = Molecule('3PTB')
>>> tryp_op, df = systemPrepare(tryp, return_details=True)
>>> tryp_op.write('/tmp/3PTB_prepared.pdb')
>>> df.to_excel("/tmp/tryp-report.csv")
>>> df # doctest: +NORMALIZE_WHITESPACE
resname protonation resid insertion chain segid pKa buried
0 ILE ILE 16 A 0 7.413075 0.839286
1 VAL VAL 17 A 0 NaN NaN
2 GLY GLY 18 A 0 NaN NaN
3 GLY GLY 19 A 0 NaN NaN
4 TYR TYR 20 A 0 9.590845 0.146429
.. ... ... ... ... ... ... ... ...
282 HOH WAT 804 A 1 NaN NaN
283 HOH WAT 805 A 1 NaN NaN
284 HOH WAT 807 A 1 NaN NaN
285 HOH WAT 808 A 1 NaN NaN
286 HOH WAT 809 A 1 NaN NaN
[287 rows x 8 columns]
>>> tryp_op = systemPrepare(tryp, pH=1.0)
>>> tryp_op.write('/tmp/3PTB_pH1.pdb')
The following will force the preparation to freeze residues 36 and 49 in place
>>> tryp_op = systemPrepare(tryp, no_opt=["protein and resid 36", "chain A and resid 49"])
The following will disable protonation on residue 32 of the protein
>>> tryp_op = systemPrepare(tryp, no_prot=["protein and resid 32",])
The following will disable titration and protonation on residue 32
>>> tryp_op = systemPrepare(tryp, no_titr=["protein and resid 32",], no_prot=["protein and resid 32",])
The following will force residue 40 protonation to HIE and 57 to HIP
>>> tryp_op = systemPrepare(tryp, force_protonation=[("protein and resid 40", "HIE"), ("protein and resid 57", "HIP")])
systemPrepare() is a convenience function. Using it is not
mandatory. You can manipulate the input molecule with your custom
functions. In particular,
Addition of hydrogen atoms is not required
Protonation states are set by renaming residues
HIS and other residues can be edited as coordinates
Example#
Prepare trypsin (PDB: 3PTB) at pH 7.
tryp = Molecule("3PTB")
tryp_op = systemPrepare(tryp)
2021-11-16 09:54:22,642 - moleculekit.readers - INFO - Using local copy for 3PTB: /home/sdoerr/Work/moleculekit/moleculekit/test-data/pdb/3ptb.pdb
2021-11-16 09:54:22,818 - moleculekit.tools.preparation - WARNING - Both chains and segments are defined in Molecule.chain / Molecule.segid, however they are inconsistent. Protein preparation will use the chain information.
---- Molecule chain report ----
Chain A:
First residue: ILE:16:
Final residue: HOH:809:
---- End of chain report ----
2021-11-16 09:54:24,870 - moleculekit.tools.preparation - WARNING - The following residues have not been optimized: BEN, CA
2021-11-16 09:54:24,964 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:22 to CYX
2021-11-16 09:54:24,965 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:40 to HIE
2021-11-16 09:54:24,965 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:42 to CYX
2021-11-16 09:54:24,965 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:57 to HIP
2021-11-16 09:54:24,966 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:58 to CYX
2021-11-16 09:54:24,966 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:91 to HID
2021-11-16 09:54:24,966 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:128 to CYX
2021-11-16 09:54:24,967 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:136 to CYX
2021-11-16 09:54:24,967 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:157 to CYX
2021-11-16 09:54:24,968 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:168 to CYX
2021-11-16 09:54:24,968 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:182 to CYX
2021-11-16 09:54:24,968 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:191 to CYX
2021-11-16 09:54:24,969 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:201 to CYX
2021-11-16 09:54:24,969 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:220 to CYX
2021-11-16 09:54:24,969 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:232 to CYX
2021-11-16 09:54:24,972 - moleculekit.tools.preparation - WARNING - Dubious protonation state: the pKa of 3 residues is within 1.0 units of pH 7.4.
2021-11-16 09:54:24,973 - moleculekit.tools.preparation - WARNING - Dubious protonation state: ILE 16 A (pKa= 7.41)
2021-11-16 09:54:24,973 - moleculekit.tools.preparation - WARNING - Dubious protonation state: TYR 39 A (pKa= 8.24)
2021-11-16 09:54:24,974 - moleculekit.tools.preparation - WARNING - Dubious protonation state: HIS 57 A (pKa= 7.44)
Visualize protonation of residue 40#
tryp_op.view(style="Licorice",sel="resid 40",hold=True)
tryp_op.view(style="Lines",sel="same residue as exwithin 4 of resid 40")
A Jupyter Widget
Preparation report#
If the return_details argument is set, an object of type
pandas.DataFrame is returned as a second return value. It
carries a wealth of information on the preparation results.
tryp_op, df = systemPrepare(tryp, return_details=True)
df
2021-11-16 09:54:27,991 - moleculekit.tools.preparation - WARNING - Both chains and segments are defined in Molecule.chain / Molecule.segid, however they are inconsistent. Protein preparation will use the chain information.
---- Molecule chain report ----
Chain A:
First residue: ILE:16:
Final residue: HOH:809:
---- End of chain report ----
2021-11-16 09:54:30,079 - moleculekit.tools.preparation - WARNING - The following residues have not been optimized: BEN, CA
2021-11-16 09:54:30,177 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:22 to CYX
2021-11-16 09:54:30,177 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:40 to HIE
2021-11-16 09:54:30,178 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:42 to CYX
2021-11-16 09:54:30,178 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:57 to HIP
2021-11-16 09:54:30,178 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:58 to CYX
2021-11-16 09:54:30,179 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:91 to HID
2021-11-16 09:54:30,179 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:128 to CYX
2021-11-16 09:54:30,179 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:136 to CYX
2021-11-16 09:54:30,180 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:157 to CYX
2021-11-16 09:54:30,180 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:168 to CYX
2021-11-16 09:54:30,181 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:182 to CYX
2021-11-16 09:54:30,182 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:191 to CYX
2021-11-16 09:54:30,182 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:201 to CYX
2021-11-16 09:54:30,183 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:220 to CYX
2021-11-16 09:54:30,183 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:232 to CYX
2021-11-16 09:54:30,188 - moleculekit.tools.preparation - WARNING - Dubious protonation state: the pKa of 3 residues is within 1.0 units of pH 7.4.
2021-11-16 09:54:30,188 - moleculekit.tools.preparation - WARNING - Dubious protonation state: ILE 16 A (pKa= 7.41)
2021-11-16 09:54:30,189 - moleculekit.tools.preparation - WARNING - Dubious protonation state: TYR 39 A (pKa= 8.24)
2021-11-16 09:54:30,189 - moleculekit.tools.preparation - WARNING - Dubious protonation state: HIS 57 A (pKa= 7.44)
| resname | protonation | resid | insertion | chain | segid | pKa | buried | |
|---|---|---|---|---|---|---|---|---|
| 0 | ILE | ILE | 16 | A | 0 | 7.413075 | 0.839286 | |
| 1 | VAL | VAL | 17 | A | 0 | NaN | NaN | |
| 2 | GLY | GLY | 18 | A | 0 | NaN | NaN | |
| 3 | GLY | GLY | 19 | A | 0 | NaN | NaN | |
| 4 | TYR | TYR | 20 | A | 0 | 9.590845 | 0.146429 | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 282 | HOH | WAT | 804 | A | 1 | NaN | NaN | |
| 283 | HOH | WAT | 805 | A | 1 | NaN | NaN | |
| 284 | HOH | WAT | 807 | A | 1 | NaN | NaN | |
| 285 | HOH | WAT | 808 | A | 1 | NaN | NaN | |
| 286 | HOH | WAT | 809 | A | 1 | NaN | NaN |
287 rows × 8 columns
Most of it is accessible in the data property, as a pandas
DataFrame.
df.columns
Index(['resname', 'protonation', 'resid', 'insertion', 'chain', 'segid', 'pKa',
'buried'],
dtype='object')
df.loc[:,['resname','resid','pKa','protonation']].head(10)
| resname | resid | pKa | protonation | |
|---|---|---|---|---|
| 0 | ILE | 16 | 7.413075 | ILE |
| 1 | VAL | 17 | NaN | VAL |
| 2 | GLY | 18 | NaN | GLY |
| 3 | GLY | 19 | NaN | GLY |
| 4 | TYR | 20 | 9.590845 | TYR |
| 5 | THR | 21 | NaN | THR |
| 6 | CYS | 22 | 99.990000 | CYX |
| 7 | GLY | 23 | NaN | GLY |
| 8 | ALA | 24 | NaN | ALA |
| 9 | ASN | 25 | NaN | ASN |
As such, it can be easily queried and written as a spreadsheet in Excel or CSV format.
df.to_csv("./tryp_data.csv")
Special case: Membrane proteins#
Membrane-embedded proteins are in contact with an hydrophobic region which may alter pKa values for membrane-exposed residues (image taken from Teixeira et al., J. Chem. Theory Comput., 2016, 12 (3), pp 930–934).
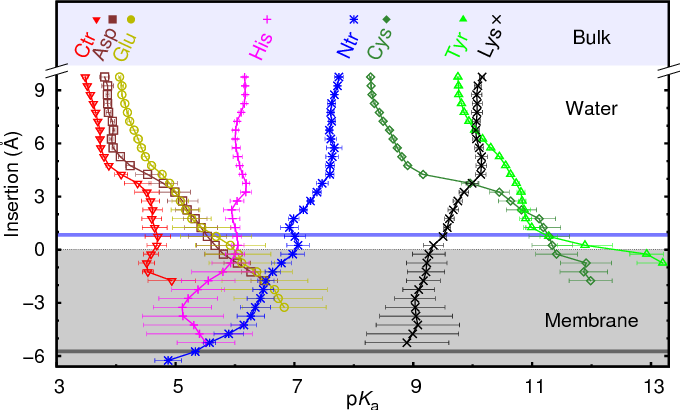
pka_membranes#
Although the effect is not currently taken into account quantitatively,
if a hydrophobic_thickness argument is provided, warnings will be
generated for residues exposed to the lipid region.
The following example shows the preparation of the mu opioid receptor, 4DKL. The pre-oriented structure is retrieved from the OPM database.
from moleculekit.util import opm
mor, thickness = opm("4dkl")
print(thickness)
mor.filter("protein and noh")
mor_opt, df = systemPrepare(mor, return_details=True,
hydrophobic_thickness=thickness)
df.to_csv("./mor_exposed_residues.csv")
2021-11-16 09:55:02,479 - moleculekit.molecule - INFO - Removed 2546 atoms. 4836 atoms remaining in the molecule.
2021-11-16 09:55:02,510 - moleculekit.molecule - INFO - Removed 364 atoms. 4472 atoms remaining in the molecule.
32.0
---- Molecule chain report ----
Chain A:
First residue: MET:65:
Final residue: ILE:352:
Chain B:
First residue: MET:65:
Final residue: ILE:352:
---- End of chain report ----
2021-11-16 09:55:07,022 - moleculekit.tools.preparation - INFO - Modified residue ASP:A:114 to ASH
2021-11-16 09:55:07,023 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:140 to CYX
2021-11-16 09:55:07,024 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:171 to HID
2021-11-16 09:55:07,024 - moleculekit.tools.preparation - INFO - Modified residue CYS:A:217 to CYX
2021-11-16 09:55:07,025 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:223 to HID
2021-11-16 09:55:07,025 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:297 to HID
2021-11-16 09:55:07,025 - moleculekit.tools.preparation - INFO - Modified residue HIS:A:319 to HIE
2021-11-16 09:55:07,026 - moleculekit.tools.preparation - INFO - Modified residue ASP:B:114 to ASH
2021-11-16 09:55:07,026 - moleculekit.tools.preparation - INFO - Modified residue CYS:B:140 to CYX
2021-11-16 09:55:07,027 - moleculekit.tools.preparation - INFO - Modified residue HIS:B:171 to HID
2021-11-16 09:55:07,027 - moleculekit.tools.preparation - INFO - Modified residue CYS:B:217 to CYX
2021-11-16 09:55:07,027 - moleculekit.tools.preparation - INFO - Modified residue HIS:B:223 to HID
2021-11-16 09:55:07,027 - moleculekit.tools.preparation - INFO - Modified residue HIS:B:297 to HID
2021-11-16 09:55:07,028 - moleculekit.tools.preparation - INFO - Modified residue HIS:B:319 to HIE
2021-11-16 09:55:07,028 - moleculekit.tools.preparation - WARNING - Dubious protonation state: the pKa of 4 residues is within 1.0 units of pH 7.4.
2021-11-16 09:55:07,030 - moleculekit.tools.preparation - WARNING - Dubious protonation state: MET 65 A (pKa= 7.76)
2021-11-16 09:55:07,030 - moleculekit.tools.preparation - WARNING - Dubious protonation state: ASP 114 A (pKa= 7.85)
2021-11-16 09:55:07,030 - moleculekit.tools.preparation - WARNING - Dubious protonation state: MET 65 B (pKa= 7.76)
2021-11-16 09:55:07,031 - moleculekit.tools.preparation - WARNING - Dubious protonation state: ASP 114 B (pKa= 7.85)
2021-11-16 09:55:07,037 - moleculekit.tools.preparation - WARNING - Predictions for 18 residues may be incorrect because they are exposed to the membrane (-16.0<z<16.00 and buried<0.8%).
Case 2. Building with a ligand#
Coexistence and automatic placement of a ligand requires further manipulation, because:
The ligand may have to be arranged in a geometrically sensible way
We likely need additional parameters and topologies
See the tutorial System Building Trypsin-Benzamidine.
Case 3. Membrane proteins#
Pre-equilibrated membranes can be merged with pre-oriented systems, e.g. downloaded from the OPM. See the tutorial System Building μ-opioid Receptor in Membrane.
Citations#
Please acknowledge your use of PDB2PQR and PropKa by citing:
Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res, 35, W522-5, 2007.
Sondergaard, Chresten R., Mats HM Olsson, Michal Rostkowski, and Jan H. Jensen. “Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values.” Journal of Chemical Theory and Computation 7, no. 7 (2011): 2284-2295.