
Ligand binding MSM: trypsin + benzamidine#
You will learn: how to take unbiased binding trajectories of a small drug-like ligand and a protein receptor and build a Markov-state model that gives you the bound macrostate, the binding pathways, the binding affinity (Kd), and on / off rates.
Prerequisites:
HTMD installed.
wgetavailable on$PATH.~5 GB of disk for the trajectory download.
The system#
Trypsin is a serine protease; benzamidine is a small competitive inhibitor that docks into the S1 specificity pocket. The dataset contains short unbiased trajectories started from many random ligand poses around the protein - some find the binding pocket, most don’t. Aggregate sampling is ≈ 17 µs across 852 trajectories.
The point of this analysis is to reconstruct the binding free-energy surface and rates without ever steering the ligand - just by counting transitions between metastable states discovered by clustering the trajectories.
The flow#
Same MSM pipeline as the villin folding tutorial - the only thing that changes is the projection: for a binding problem the slow coordinate is the protein-ligand distance pattern, not the protein’s own contact map.
Download + simlist.
Project with
MetricDistancebetween protein Cα atoms and ligand heavy atoms.TICA to find the slow binding modes.
Cluster, build MSM, lump into macrostates.
Read out the FES, MFPTs, kon / koff, and Kd.
Setup#
import os
from glob import glob
from pathlib import Path
from htmd.ui import (
simlist, simmerge,
Metric, MetricDistance,
TICA, Model, Kinetics,
)
from sklearn.cluster import MiniBatchKMeans
Please cite HTMD: Doerr et al.(2016)JCTC,12,1845. https://dx.doi.org/10.1021/acs.jctc.6b00049
HTMD Documentation at: https://software.acellera.com/htmd/
You are on the latest HTMD version (2.8.5.dev17+gf26491d44.d20260602).
2026-06-02 18:40:37,204 - rdkit - INFO - Enabling RDKit 2026.03.2 jupyter extensions
Step 1 - Build the simlist#
The trajectory bundle ships on Figshare (HTMD tutorial data, DOI 10.6084/m9.figshare.32541291) as ligand_binding_datasets.zip (~3 GB).
The dataset is split into several “epochs” (adaptive-sampling rounds). simlist() builds one per-epoch list (duplicate folder basenames within a single call would raise), and simmerge() stitches them into a combined list and renumbers the per-sim simid indices to be sequential across the merged whole:
DATASETS = Path(os.environ["HTMD_TUTORIAL_DATASETS"]) / "ligand_binding_datasets"
topology = str(DATASETS / "1" / "filtered") # any epoch works - all share the same topology
sims = []
for epoch in sorted(DATASETS.glob("*/")):
trajs = glob(os.path.join(epoch, "filtered", "*", ""))
sims = simmerge(sims, simlist(trajs, topology))
len(sims)
852
Step 2 - Project: protein-ligand contacts#
For binding, the relevant coordinate is “which protein residue is the ligand currently in contact with”. MetricDistance computes the distance matrix between two atom selections; with metric="contacts" you get a binary contact map per frame between every protein Cα and every ligand heavy atom (1 if the distance is below the threshold, 0 otherwise - default threshold=8 Å).
metr = Metric(sims)
metr.set(MetricDistance(
"protein and name CA",
"resname MOL and noh",
periodic="selections",
metric="contacts",
))
data = metr.project()
data.fstep = 0.1
htmd.projections.metric - INFO - Frame step 0.001ns was read from the trajectories. If it looks wrong, redefine it by manually setting the MetricData.fstep property.
data.plotTrajSizes()
data.dropTraj()
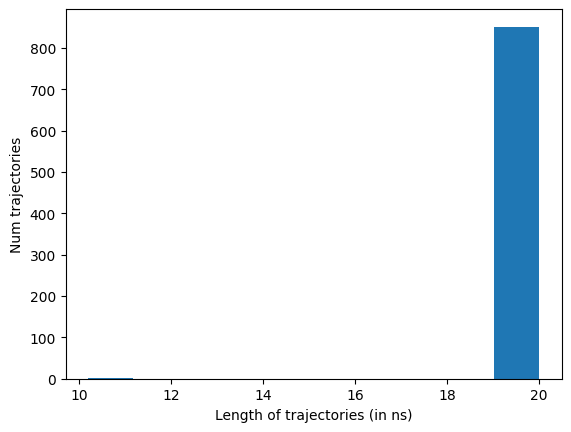
htmd.metricdata - INFO - Dropped 2 trajectories from 852 resulting in 850
array([496, 562])
periodic="selections" makes the distance calculation use minimum-image distances between the two atom selections, so a ligand that has wrapped through PBC gets compared to the closest protein image - the original coordinates aren’t modified, only the distances are computed correctly across the box.
Step 3 - TICA#
tica = TICA(data, 2, units="ns")
dataTica = tica.project(3)
Step 4 - Cluster + MSM#
dataBoot = dataTica.bootstrap(0.8)
dataBoot.cluster(MiniBatchKMeans(n_clusters=1000))
model = Model(dataBoot)
model.plotTimescales(maxlag=15, units="ns")
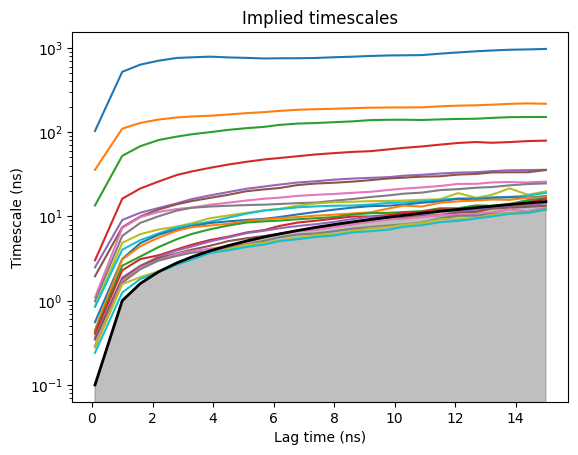
The slow timescale for binding is much shorter than folding (ligand diffusion happens on the ns-100ns scale, not µs). Read the lag off the ITS plot - usually around 5 ns for this system.
model.markovModel(5, 5, units="ns")
htmd.model - INFO - 100.0% of the data was used
htmd.model - INFO - Number of trajectories that visited each macrostate:
htmd.model - INFO - [153 105 376 127 220]
Five macrostates: bound + a few “encountered but not docked” intermediates + the bulk solution.
Step 5 - Free-energy surface#
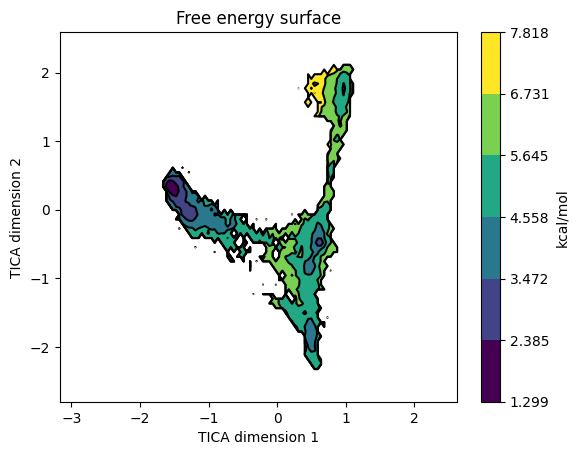
model.plotFES(0, 1, temperature=298)
model.plotFES(0, 1, temperature=298, states=True)
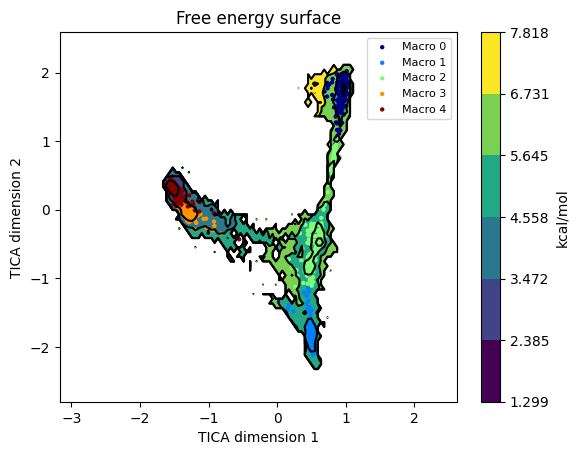
The bound state usually appears as a deep basin in one corner of TIC1 / TIC2; the bulk-solution state spreads across most of the projected area; the intermediates are shallow basins between them.
To overlay representative protein + ligand snapshots from each macrostate, run model.viewStates(ligand="resname MOL and noh") from an interactive session - it launches VMD. (Omitted here because it needs a display.)
Step 6 - Kinetics + Kd#
kin = Kinetics(model, temperature=298, concentration=0.0037)
htmd.kinetics - INFO - Detecting source state...
htmd.kinetics - INFO - Guessing the source state as the state with minimum contacts.
htmd.kinetics - INFO - Source macro = 2
htmd.kinetics - INFO - Detecting sink state...
htmd.kinetics - INFO - Sink macro = 4
concentration=0.0037 mol/L is the effective bulk ligand concentration in this simulation box (one ligand in the periodic box of this volume). Kinetics uses it to convert the simulated rates into experimental units - kon (M⁻¹ s⁻¹), koff (s⁻¹), and Kd = koff / kon.
r = kin.getRates()
print(r)
mfpton = 7.88E+02 (ns)
mfptoff = 6.80E+03 (ns)
kon = 3.43E+08 (1/M 1/s)
koff = 1.47E+05 (1/s)
koff/kon = 4.29E-04 (M)
kdeq = 3.58E-04 (M)
g0eq = -4.70 (kcal/M)
htmd.kinetics - INFO - Calculating rates between source: [np.int64(2)] and sink: [np.int64(4)] states.
htmd.kinetics - INFO - Concentration correction of -3.32 kcal/mol.
getRates() returns a single Rates object for the auto-detected source → sink pair (bulk → bound here). It carries the MFPT, rate, and kon / koff / Kd. Match against the experimental Kd to validate the model. Use kin.plotRates() (below) for an all-pairs visualisation, or call getRates(source=..., sink=...) explicitly for other pairings.
kin.plotRates()
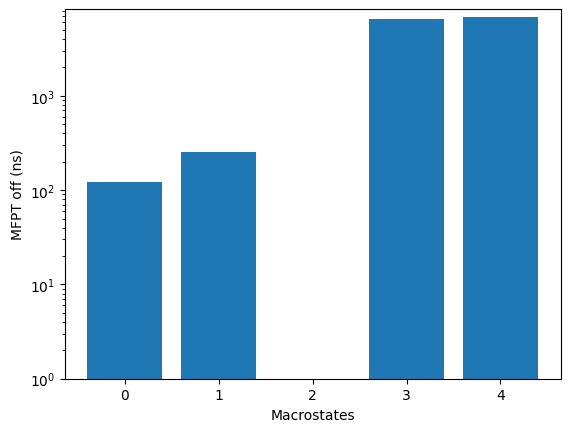
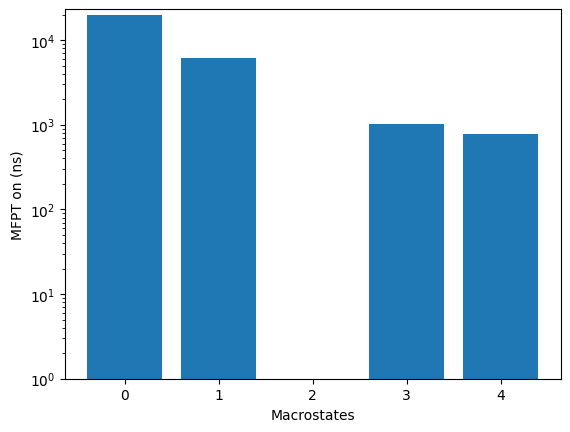
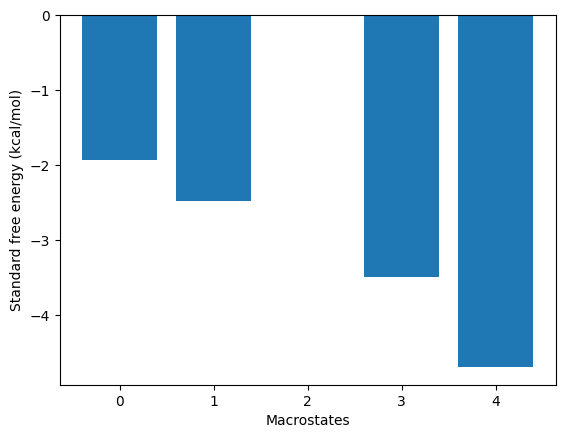
kin.plotFluxPathways()
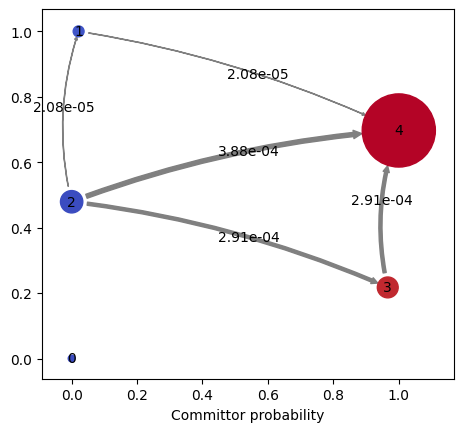
Path flux %path %of total path
0.00038753881932143707 55.4% 55.4% [2 4]
0.00029093136484922627 41.6% 97.0% [2 3 4]
2.0777315285460142e-05 3.0% 100.0% [2 1 4]
The flux pathways show which intermediates the ligand visits on the way from solution to the bound pocket - useful for understanding the binding mechanism (e.g. is there a kinetic trap on the surface? Does the ligand approach from a specific direction?).
Parameters that matter#
Knob |
Effect |
|---|---|
|
Coarse but the right default for binding - thresholding collapses the huge unbound bulk region into a single “no contacts” state. |
|
Default 8 Å. Lower (e.g. 5 Å) tightens what counts as “in contact” and emphasises tighter poses; higher dilates the bound basin. |
|
Critical for kon and Kd. Compute it as (nligands / nwaters) · 55.4 mol/L - the water count tracks the real bulk volume more accurately than the box volume, which over-counts because it includes the protein’s excluded volume. |
|
Essential when the ligand wraps through the box during the trajectory. Skipping it produces nonsense contacts at PBC crossings. |
|
More macrostates → more pathway resolution but harder to interpret. 4-6 is typical for binding. |
Gotchas#
The bulk state is huge and unstructured. With
metric="contacts"the unbound bulk collapses to essentially a single microstate (all-zero contact vector), which is exactly what you want for binding analysis. Don’t over-interpret intermediate basins that have very small populations - they may be undersampled.Bound-state validation. Before trusting Kd, open
viewStates(ligand=...)and confirm the bound macrostate actually puts the ligand in the experimental pocket. If it’s binding to the wrong site, your model is sampling a metastable mis-pose.Symmetric ligands. Benzamidine is roughly C₂v-symmetric; for ligands without that symmetry, atom-pair distances can flip when the ligand rotates 180°. Either symmetrise the contact features manually or accept that the model will distinguish the two flipped orientations as separate states.
See also#
MSM workflow explanation - what’s happening under the hood.
Protein folding MSM - same pipeline, different projection.
Adaptive sampling - how the binding trajectory set was generated (random starting poses + adaptive sampling targeting under-explored regions).