
NNP/MM simulations#
Recent developments in machine learning have enabled the use of neural networks to estimate the potential energy of molecules at much higher accuracy than classical force fields. This is due to the fact that neural networks can capture complex non-linear dependencies in the potential energy surface and are directly trained on high-quality quantum mechanics (QM) data.
ACEMD supports running molecular dynamics simulations with neural networks in so-called hybrid NNP/MM simulations [JChemInfModel2023]. In these simulations a part of the system (usually a small molecule, like a ligand) is described with a neural network potential, while the rest of the system is described with a classical force field. Non-bonded interactions between the small molecule and the rest of the system are described with classical force field parameters.
To use NNP/MM simulations with ACEMD one needs a classically built system (AMBER or CHARMM) including all atoms of the simulation.
You can download here the example input file for an NNP/MM simulation of benzamidine in solvent.
In this simulation the benzamidine molecule is described with a neural network potential while the rest of the system (in this case just the waters)
is described with the AMBER force field.
The input file of ACEMD can then be configured as follows to run a NNP/MM simulation (see Neural Network Potentials for more details):
barostat: true
thermostat: true
thermostattemperature: 300
velocities: 300
structure: structure.prmtop
coordinates: structure.pdb
boxsize: [22.66, 25.397, 25.982]
minimize: 500
timestep: 2
run: 2ns
nnp:
file: aceff_v1.0.ckpt
name: TorchMD-Net
sel: "resname BEN"
type: torch
This instructs ACEMD to simulate the residue with name BEN using the neural network potential stored in the file aceff_v1.0.ckpt
while the rest of the system and its intramolecular interactions with BEN are described with the classical force field parameters
read from the structure.prmtop file.
The user should make sure that the atom selection in sel encompasses the whole molecule of interest and doesn’t miss atoms
covalently bonded to it.
A timestep of 2 fs is recommended for NNP/MM simulations as NNP/MM simulations are not very stable at larger timesteps.
Obtaining AceFF NNP models#
To run NNP/MM simulations with ACEMD you need to obtain a NNP model.
AceFF is the state-of-the-art family of neural network potentials developed by Acellera. They and can be downloaded from the AceFF HuggingFace page.
First register to HuggingFace if you haven’t already and then request access to the models from the above webpage.
Activate your ACEMD conda environment and login into your HuggingFace account from the command line:
huggingface-cli login
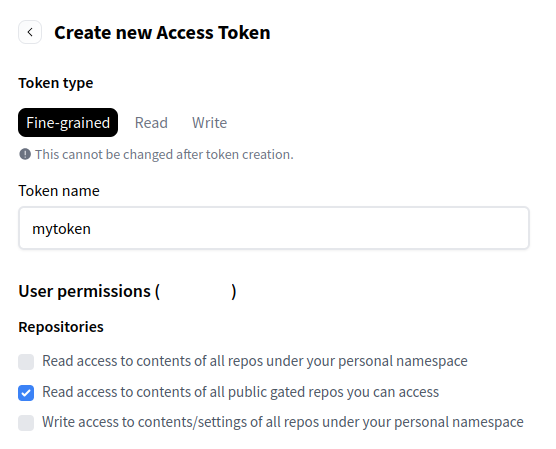
To login you will need to generate an access token from your HuggingFace account. Take care to give permission for the token to “Read access to contents of all public gated repos you can access” as in the following screenshot:
Once you have been granted access, download the desired model with the following command:
huggingface-cli download Acellera/AceFF-1.0 aceff_v1.0.ckpt --local-dir .
Warning
Currently AceFF 1.0 models can only describe molecules with a total charge of 0, 1 or -1. Molecules with larger total charge will not be described correctly by the NNP and the simulation will likely be inaccurate. Upcoming versions of AceFF are meant to tackle this issue.
Running the simulation#
Finally, you can execute the simulation with a simple acemd call:
acemd
Using the ANI family of neural network potentials#
ACEMD also supports running NNP/MM simulations with the ANI family of neural network potentials [ANI2020_1], [ANI2020_2]. The ANI NNPs were some of the first publicly available neural network potentials and are still used today as benchmark potentials due to their accuracy, stability and efficiency.
Warning
The ANI models cannot correctly describe molecules with total charge different than 0. You are advised to instead use the AceFF models which can correctly describe charged molecules.
You can use the ANI potentials in ACEMD by setting up the input config file as follows:
barostat: true
thermostat: true
thermostattemperature: 300
velocities: 300
structure: structure.prmtop
coordinates: structure.pdb
boxsize: [22.66, 25.397, 25.982]
minimize: 500
timestep: 2
run: 2ns
nnp:
name: ANI-2x
sel: "resname BEN"
type: torch
It is not necessary to specify the file parameter as it will be automatically downloaded when an ANI model is selected.
Galvelis et al., J. Chem. Inf. Model., 2023 Sep 25;63(18):5701-5708, https://doi.org/10.1021/acs.jcim.3c00773
Smith, J.S. et al. Sci. Data 7, 134 (2020). https://doi.org/10.1038/s41597-020-0473-z
Devereux et al., J. Chem. Theory Comput., 2020, 16, 7, 4192-4202, https://doi.org/10.1021/acs.jctc.0c00121